
清澤のコメント:京都大学iPS細胞研究所のプレスリリースによると、共同研究グループは、患者さん由来の人工多能性幹細胞(iPS細胞)から3次元網膜オルガノイドを作製。またゼブラフィッシュeys変異を作製して解析することにより、光刺激による視細胞の細胞死がEYS 関連網膜変性疾患の病態に重要な役割を果たしていることを発見したとのことである。その内容を克明に理解する事は難しいが、日本でも最も5から30%と最も普遍的な種類の網膜色素変性の発症予防法が、既に広く行われて基地たことではあるが、光毒性の回避であると解ったことは間違いなさそうです。プレスリリースの要点と原著論文の要旨および導入部分を採録します。
ーーーーープレスリリースからーーーーー
本研究成果は、未知であったEYS 関連網膜変性疾患の病態メカニズムを明らかにするとともに、特定の波長光への暴露を遮断することが治療の選択肢の一つになる可能性を示唆している。
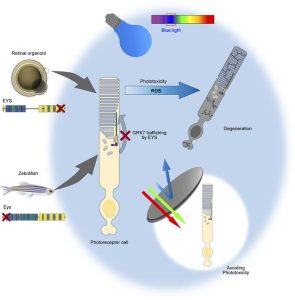
遺伝性網膜変性疾患(Inherited retinal dystrophies:IRD)の最も多い原因としてEyes shut homolog(EYS )遺伝子の変異が知られていたが、EYS は哺乳類における研究モデルがないという課題があった。今回、共同研究グループは、健常者と網膜変性疾患の患者それぞれのiPS細胞から網膜オルガノイドを作製し、病態解析を行った。健常者由来オルガノイドの視細胞においてEYSタンパク質は結合線毛や外節領域に局在していたのに対し、患者由来オルガノイドではこれらの領域での局在量が低下し、細胞質内に局在異常を生じていた。EYSは視細胞外節で働くタンパクの一つであるG-protein-coupled receptor kinase(GRK7)という分子と直接結合し、その外節への輸送に関与していることを発見し、患者由来オルガノイドでは外節へのGRK7の輸送量が低下していることを見いだした。また、患者由来オルガノイドでは光刺激後に視細胞の細胞死が誘導されることが分かった。さらに、ゼブラフィッシュeys 遺伝子変異体を作製し解析を行い、ゼブラフィッシュモデルにおいても光刺激により視細胞の細胞死が誘導されることを明らかにしたという。
この研究成果は、2024年4月22日(米国時間)に科学雑誌「JCI Insight」オンライン版に掲載された。
――――この論文の要旨と導入部分を採録します―――――オープンアクセス | 10.1172/jci.insight.174179
光毒性の回避は、EYS機能不全によって引き起こされる網膜ジストロフィーに対する潜在的な治療アプローチである
大塚祐樹、ほか1,2,3 2024 年 4 月 22 日公開 – 詳細情報
抄録:
遺伝性網膜ジストロフィー (IRD) は、視力喪失につながる進行性の疾患です。アイズシャットホモログ ( EYS ) 遺伝子の変異は、IRD の最も頻繁な原因の 1 つです。しかし、変異型 EYS による光受容細胞変性のメカニズムは完全には解明されていません。今回、我々はEYS関連網膜ジストロフィー(EYS -RD)患者由来の人工多能性幹細胞(iPSC)から網膜オルガノイドを作製した。 RDオルガノイドの光受容細胞では、EYSと光毒性を担うタンパク質の1つであるGタンパク質共役型受容体キナーゼ7(GRK7)の両方が、生理学的に存在する外節に存在しなかった。さらに、RD オルガノイドの光受容細胞は光刺激、特に青色光に対して脆弱でした。eysノックアウトゼブラフィッシュでも観察されたGRK7の誤った局在化は、対照EYSをRDオルガノイドの光受容細胞に送達することによって逆転した。これらの発見は、光毒性を回避することがEYS -RDの潜在的な治療法であることを示唆しています。
遺伝性網膜ジストロフィー(IRD)は、不可逆的な視力喪失につながる進行性の光受容体細胞死を特徴としています。これらは視覚障害や失明の主な原因であり、世界中で 450 万人以上が罹患しています。過去 10 年間に大きな進歩があったにもかかわらず、IRD の治療は非常に困難であり、克服すべき課題はまだ多くあります。現在までに、250 を超える遺伝子の変異が IRD (網膜情報ネットワーク [RetNet]、https://sph.uth.edu/retnet/ ) を引き起こすことが確認されています。
アイズ・シャット・ホモログ ( EYS ) 遺伝子は、2008 年に網膜色素変性症 (RP) の常染色体劣性原因遺伝子として初めて同定されました。EYS は主に網膜、特に光受容細胞で発現します (Human Protein Atlas; https://www.proteinatlas.org/ENSG00000188107-EYS/tissue )。EYS遺伝子の変異は、RP の最も頻繁な原因の 1 つであり、その罹患率は約 5% ~ 30% であると報告されています。 EYSは染色体 6p12 上に位置し、2 Mb のゲノム DNA にわたっています。全長のヒトEYS転写物は 44 個のエクソンで構成され、5 個のラミニン AG 様ドメインと 28 個の上皮成長因子 (EGF) または EGF 様ドメインで構成される 3,165 アミノ酸のタンパク質をコードしています。 EYS は人間の目に発現する最大の分子の 1 つであり、そのサイズにより基礎研究と遺伝子治療の両方において厳しい制限が生じています。
EYS は、最も一般的な動物モデルであるマウス、ラット、モルモットを含むいくつかの哺乳類系統のゲノムで欠損または中断されており、このことがEYS関連網膜ジストロフィー ( EYS -RD)の調査を困難にしています。したがって、ゼブラフィッシュは、Eys タンパク質の役割を研究するための代替脊椎動物モデルとして使用されています。これらの研究によると、機能喪失がEYS -RD の根底にある分子機構であると考えられています、Eys は形態構造の維持に関与すると予想されています。光受容体または外節 (OS) へのタンパク質輸送。一方、変異体の位置はEYS -RD 患者の表現型と疾患の重症度に影響を与える傾向があり、追加のメカニズムが存在する可能性があることが示唆されています。 2つの家系におけるEYS -RDの病理組織像を記載した報告は 1 つありますが(家系 1 の c.2259+1G>A および c.2620C>T、家系 2 の c.4350_4356del および c.2739_3244del)、調査された症例は病気が進行した段階にあり、光受容体層はすでに高度に組織化されていませんでした。したがって、EYS の正確な分子機能と、変異体 EYS がどのように網膜変性を引き起こすのかはまだ解明されていません。
EYSの機能とIRDの病因における変異型EYSの関与をより深く理解するには、ヒトサンプルを活用することが不可欠です。ヒト人工多能性幹細胞 (iPSC) により、さまざまな疾患を研究するためのヒト臓器特異的細胞や in vitro モデルを生成できるようになりました。網膜に関しては、緑内障、レーベル先天性黒内障、シュタルガルト病、および X 連鎖若年性網膜剥離で3次元網膜オルガノイドを作製する方法が開発されています。特に、 RP2 、 RPGR 、 USH2A などのいくつかの RP 原因遺伝子の病因が、患者由来のオルガノイドを用いて研究され、疾患の表現型が再現されています。これらの研究は、ヒト iPSC が IRD 関連の分子病因の研究に有望なモデルを提供することを実証しました。
EYS -RDの病因を調べるために、患者由来の iPSC から生成された網膜オルガノイドが分析されました。これは、EYS の機能とその病因の分子機構を研究するためにヒト iPSC が使用されているという、我々の知る限りでは初めての報告です。今回の研究では、ヒトEYSは光受容細胞の連結繊毛(CC)とOSに局在していたが、変異体切断型EYSはこれらの領域には分布していなかった。私たちは、EYS が OS タンパク質である G タンパク質共役受容体キナーゼ 7 (GRK7) の輸送に役割を果たしていることを発見しました。GRK7 は EYS とともに進化的に保存されており、光応答の回復に関与しています。 EYS -RD オルガノイドとゼブラフィッシュは、OS における GRK7 欠乏に関連する光誘発性光受容体細胞死を示しました。さらに、RD オルガノイドにおける光誘発細胞死は、特に青色光によって引き起こされました。



コメント