
 OPA1遺伝子に新規de novo変異が見出された両眼視神経萎縮の男児例:が日本眼科学会雑誌に収載されている。この症例報告では、常染色体顕性遺伝(優性遺伝)視神経萎縮(DOA)が OPA1遺伝子変異により生じているが、それは両親にその遺伝子のないde novo変異であったという例である。
OPA1遺伝子に新規de novo変異が見出された両眼視神経萎縮の男児例:が日本眼科学会雑誌に収載されている。この症例報告では、常染色体顕性遺伝(優性遺伝)視神経萎縮(DOA)が OPA1遺伝子変異により生じているが、それは両親にその遺伝子のないde novo変異であったという例である。 遺伝性視神経症, 常染色体顕性遺伝(優性遺伝)視神経萎縮(DOA), OPA1遺伝子変異, de novo変異, 全エクソーム解析(WES)
目 的:家族歴のない原因不明の両眼視神経萎縮に対して実施した全エクソーム解析(WES)により,常染色体顕性遺伝(優性遺伝)視神経萎縮(DOA)の原因であるOPA1遺伝子に新規変異が見出された男児例について報告する.
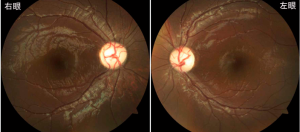
症 例:7歳,男児.就学時健診で両眼の視力障害を指摘され,精査のため当院を受診した.視力障害の自覚および家族歴はなかった.矯正視力は右0.4,左0.5であった.前眼部および中間透光体に異常はなく,眼底所見として両眼の視神経乳頭蒼白を認めた.光干渉断層計検査では黄斑部網膜外層に構造異常はみられなかったが,網膜内層厚および視神経乳頭周囲網膜神経線維層厚が有意に菲薄化していた.視神経萎縮の原因を明らかにする目的で,トリオ(患児および両親)サンプルに対してWESを施行し,イントロン27のドナースプライシング部位に新規変異(c.2818+4A>G)をヘテロ接合で認めた.両親は同変異を有しておらず,de novo変異と同定された.18歳時の矯正視力は右0.2,左0.1となり,Goldmann視野検査では両眼に盲点中心暗点がみられた.臨床像はDOAに合致しており,11年の観察期間中,緩徐であったが進行性の視力障害に加え,近視化を認めた.
結 論:DOAに合致する臨床所見がみられれば,常染色体顕性遺伝(優性遺伝)性が否定されてもトリオサンプルの遺伝学的検査により,OPA1遺伝子にde novo変異が検出されるケースが存在する.(日眼会誌126:983-990,2022)
緒 言
常染色体顕性遺伝(優性遺伝)視神経萎縮(autosomal dominant optic atrophy:DOA)は,学童期の視力障害の原因として鑑別にあげるべき疾患の一つである.1959年にKjerが多数家系を報告したことから,Kjerタイプ視神経萎縮とも呼ばれている.日本人における有病率は分かっていないが,欧米では5万人に1人と推定されている.2000年,DOAがOPA1遺伝子のヘテロ接合変異によって発症することが報告された.OPA1遺伝子(NM_015560.3)は,3番染色体長腕(3q29)に局在し,約105 kb(105,000塩基対)からなる大きな遺伝子であり,29個のエクソンから構成されている.2002年,ShimizuらがOPA1遺伝子変異の日本人DOA家系を本邦で初めて報告した.また,これまでの研究から,遺伝子変異のパターンは,OPA1蛋白質が生成されても短縮するような短縮型変異が多いことが判明している.DOAは常染色体顕性遺伝(優性遺伝)性疾患であるため,両親のどちらかの遺伝子変異が子に遺伝することによって発症する.一方,子のゲノムDNA内に親が持たない新たな変異が生じる場合があり,de novo変異と呼ばれている.de novo変異は,配偶子形成過程のDNA複製時のエラーなどが原因となって発生すると考えられており,OPA1遺伝子関連視神経萎縮でde novo変異が検出されるケースはまれである.
今回,著者らは原因不明の両眼視神経萎縮に対して,次世代シークエンサーを用いた全エクソーム解析(whole-exome sequencing:WES)を実施し,OPA1遺伝子に新規de novo変異が見つかりOPA1遺伝子変異関連視神経萎縮と診断された症例を経験したので報告する.



コメント