
 清澤のコメント:我々も1例報告をしたことがあったアルポート症候群の眼症状をまとめた総説です。アルポート症候群患者が示す眼の症状には、円錐水晶体、黄斑フレック、網膜症、角膜の変化、黄斑菲薄化、黄斑円孔、網膜菲薄化、白内障、前水晶体嚢の変化、黄斑反射の鈍化などがあるとしています。
清澤のコメント:我々も1例報告をしたことがあったアルポート症候群の眼症状をまとめた総説です。アルポート症候群患者が示す眼の症状には、円錐水晶体、黄斑フレック、網膜症、角膜の変化、黄斑菲薄化、黄斑円孔、網膜菲薄化、白内障、前水晶体嚢の変化、黄斑反射の鈍化などがあるとしています。
ーーーーーーー
アルポート症候群の眼症状と治療の可能性:系統的レビュー;
概要
目的。アルポート症候群 (AS) は、重度でまれな遺伝性疾患であり、末期腎疾患、聴覚変性、および眼の異常につながる可能性があります。聴覚障害および腎障害に関連する AS に関する広範な研究にもかかわらず、AS の眼への症状についてはさらに研究が必要です。このシステマティック レビューは、AS 患者の一般的な眼の異常を要約し、これらの異常に対する潜在的な治療オプションを調査することを目的としています。メソッド。PubMed、MEDLINE、および EMBASE データベースは、1977 年 1 月から 2022 年 4 月まで体系的に検索されました。英語で発表され、AS 患者の眼の異常を調査した論文のみが選択されました。追加の研究のために、含まれている論文の参考文献リストを手動で検索しました。結果. 195 人の患者を含む合計 23 の記事がこのレビューに含まれていました。AS患者の一般的な眼の症状は、水晶体、黄斑円孔、斑点網膜症、および黄斑の菲薄化です。公開された文献では、非 AS 患者の眼の異常を緩和するための標準的な手術手技として白内障手術と硝子体切除術の使用が説明されていますが、手術手技は大規模な調査研究で AS 異常の解決策として評価されていないことに注意する必要があります。AS のもう 1 つの有望な治療法は、原因となるCOL4バリアントを野生型に戻すことによる遺伝子治療、またはCOL4A5を伴うX連鎖 ASのエクソンスキッピング療法です。ミューテーションの切り捨て。しかし、遺伝子治療は、この疾患の胎児および初期の発達段階で発生する変化を治療することができないままです。結論。このレビューでは、AS 患者における手術手技と遺伝子治療の有効性と安全性に関する決定的な結論は得られませんでした。光コヒーレンストモグラフィー (OCT) による眼科検査と細隙灯検査による眼の異常の認識は、眼科医が腎臓専門医や他の医師による AS の診断を支援できるため、医療分野にとって重要です。早期の診断とケアにより、失明や網膜剥離などの有害な眼の転帰のリスクを最小限に抑えることができます。
ーーーーこの論文の前文は背景してと記載されています。ーーーー
1. 背景
アルポート症候群 (AS) は、腎不全、難聴、および眼の異常を特徴とするまれな (1:5,000) 異種遺伝性疾患です。ASは遺伝し、主にコラーゲン遺伝子の1つ内の突然変異によって誘発されます。COL4A3とCOL4A4は染色体 2 に位置し、 COL4A5はX染色体に位置します 。X連鎖型の疾患が最も一般的で、全AS症例のほぼ80%を占めていますが、常染色体型の疾患は、遺伝子変異体に応じて優性または劣性になり、残りの20%を占めています。さらに、一般的に男性はより深刻な影響を受けますが、COL4A5バリアントのヘテロ接合体である女性は、男性と比較して一般的に軽度で進行が遅い症状を示します . COL4A3、COL4A4、およびCOL4A5遺伝子は、それぞれコラーゲン IV α 3、α 4、およびα 5 鎖をコードします。コラーゲン IV α 345 鎖は、目、耳、および腎臓の糸球体基底膜で顕著な構造成分として機能するネットワークを形成します。COL4A5の配列バリアントAS 患者の遺伝子はユタ大学によって登録されており、現在 807 のエントリが含まれています 。これらのバリアントの約 94.4% は病原性であると考えられており、機能的な IV 型コラーゲンの量に影響を与えるナンセンスおよびミスセンスの変化につながるヌクレオチドの挿入、欠失、スプライス部位の変化、およびアミノ酸の変化が含まれています。COL4A3およびCOL4A4遺伝子のバリアントは、ライデン オープン バリエーション データベース (LOVD) で精選されており、COL4A3の 257/579 およびCOL4A4の 222/558 が病原性である可能性が高いまたは確認されていると考えられ、 COL4A5で観察されたヌクレオチドおよびアミノ酸の変化の同様の分布を示しています。研究では、 COL4A3とCOL4A4の両方に劣性ダイジェニック ヘテロ接合変異を有する個体では識別可能な表現型がほとんどまたはまったくないことが報告されています。興味深いことに、COL4A3またはCOL4A4の劣性ヘテロ接合病原性バリアントと組み合わされた病原性COL4A5バリアントは、 COL4A5バリアントのみを保有する個人と比較した場合、より深刻なタンパク尿と関連しています。これは、AS患者で観察される表現型の変動が、部分的には、影響を受けた組織に残っている機能的なIV型コラーゲン鎖の量に起因する可能性があることを示唆しています.
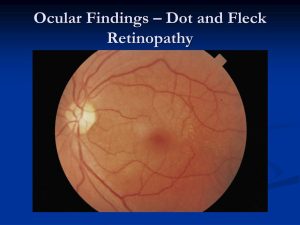
AS 患者が示す眼の症状には、水晶体、黄斑斑点、網膜症、角膜の変化、黄斑菲薄化、黄斑円孔、網膜菲薄化、白内障、前水晶体嚢の変化、黄斑反射の鈍化などがあります。レンチコヌスは、片目 (片側性) または両目 (両側性) にある前または後面の水晶体の突出として定義されます。AS 患者は通常、黄斑周囲の網膜斑点を有する可能性がある両側目の、前部レンチコーヌスを呈しますが、まれに、前部および後部のレンチコーヌスが同時に存在する場合があります。ASで一般的に観察される網膜症には、視力への影響がほとんどまたはまったくなく、治療を必要としない中心斑点および周辺斑点網膜症が含まれますが、再発性角膜びらんや後部多形性角膜ジストロフィーなどの角膜変化はあまり一般的ではありませんが、視力を損なう可能性があります . 黄斑菲薄化および網膜菲薄化は、主に画像ベースの所見であり、網膜中心部 (黄斑) または周辺網膜が薄くなり、視力に影響を与えないことを示します。手術にうまく反応せず、永久的な視力喪失につながる可能性があります。前水晶体嚢 (ALC) の異常には、水晶体を保護する ALC の裂開、破裂、または菲薄化が含まれます。最後に、白内障は水晶体から破裂した小さな部分的な裂け目が治癒した後に発生し、鈍い黄斑反射は適切な中心窩反射の喪失であり、早期発症の腎不全と関連しています。
その希少性のため、AS に対する標準的で効果的な治療法はありません。AS は一般に小児期に血尿とタンパク尿のために診断され、眼科的な理由によるものではありません。この状態は遺伝的なものであるため、多くの先行研究者が遺伝子治療が治療手段になるかどうかを調査しようと試みてきました。ただし、遺伝子治療は、疾患の胎児および初期の発達段階で発生する変化を治療しないことに注意する必要があります。さらに、遺伝子治療は、特定の遺伝病の治療に対する臨床的有用性の初期段階にまだあります。とにかく、遺伝子治療の研究とその医学への応用の可能性は有望な結果を示しており、将来的に遺伝病の治療と管理に使用できる可能性があります.
ーーーーーーーー
AS に関する以前の報告の大部分は、腎臓や耳など、目以外の領域に焦点を当てていました。しかし、コラーゲンは透明な角膜、強膜、および創傷治癒に必要なため、目の重要なタンパク質であるため、AS 患者のCOL4A3-5変異は予想通り視力低下をもたらします。X連鎖性AS患者の大規模なコホートを分析したいくつかの報告があり、水晶体、黄斑症、白内障、および眼病変の特定の眼合併症について報告されています。したがって、この系統的レビューでは、AS の眼の異常に焦点を当て、現在知られていることの要約を提供します。これには、以前の研究論文で参照されている医学的管理と潜在的な治療オプションの検討が含まれます。
ーーーーーーーーー
引用されている過去の我々のレポート:



コメント